




Computational investigation of bioactive 2,3-diaryl quinolines using DFT method: FT- IR, NMR spectra, NBO, NLO, HOMO-LUMO transitions, and quantum-chemical properties
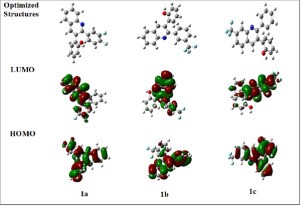
| Abstract | Quinoline derivatives are widely utilized in a variety of applications, including medicine and materials. In this work our previously developed bioactive fluorine-containing 2,3-diarylquinolines (1a, 1b and 1c) were investigated theoretically and compared with the experimental data. DFT was used to investigate the optimized geometry and geometric parameters using the B3LYP/6–311++G (d,p) basis set. DFT studies demonstrated a high degree of concordance of the predicted 1H, 13C NMR, and FT-IR spectroscopic data with the experimental results. The DFT technique was used to explore HOMO-LUMO energies, global reactive parameters, NLO, NPA, and NBO characteristics using the aforementioned level of theory and basis set. HOMO-LUMO analysis revealed that compound 1c has a slightly narrower energy gap (4.3355 eV) than compounds 1a (4.4626 eV) and 1b (4.4645 eV) suggesting all three compounds have significantly comparable NLO properties. The reported compounds have polarizability in the range 40.17 - 42.53 × 10−24 esu. Strong delocalization interactions between Lewis bonding and antibonding orbitals have been confirmed by NBO analysis. |
| Faculty |
Harminder Kaur
|
|
hkaur@pec.edu.in
|
|
| Collaborations | Govt. Home Science College, Sri Guru Teg Bahadur Khalsa College Sri Anandpur Sahib, Dr. B. R. Ambedkar National Institute of Technology Jalandhar |
| More Information | DOI: (https://doi.org/10.1016/j.molstruc.2021.132285) |